Introduction:
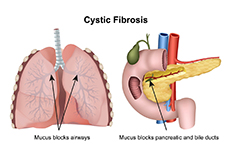
Cystic fibrosis is a hereditary disease due to mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that affects multiple organs such as the lungs, pancreas and intestine and is characterized by the production of thick and sticky mucus that causes obstruction, fibrosis and cyst formation. It is a chronic disease and prevalence is higher in Caucasians. There are over 30,000 cystic fibrosis patients in the US and incidence is 1 in 2,500-3,500 newborns. Cystic fibrosis basically affects the exocrine glands, disturbing normal functions resulting in a number of complications such as respiratory diseases, maldigestion, malabsorption and pancreatic insufficiency. It is an autosomal recessive disorder, meaning a patient would have inherited two copies of the mutated gene; one from each parent. People who inherit only one gene are “carriers” and do not show signs and symptoms. Cystic fibrosis affects mucous and sweat glands and acinar cells of the pancreas. Under normal conditions, these secretions are thin and serve as lubricants and the pancreatic enzymes produced by the acinar cells help digest food. But in cystic fibrosis mucous becomes thick, builds up and causes obstruction which stimulates fibrosis and formation of cysts. The CFTR gene is responsible for encoding “the cystic fibrosis transmembrane conductance regulator protein” which in turn is responsible for ion (chloride) and water transport across membranes. The mutation results in impairment of the chloride channels resulting in decreased chloride efflux and increased sodium and water reabsorption causing the formation of thick and sticky mucous with minimal water content and excessive salt secretion in sweat. Although the prognosis has improved, patients have shortened life expectancy. The complications include pancreatitis, diabetes mellitus, respiratory failure, cirrhosis of the liver and re-current infections. In case of lung involvement, the upward beating of cilia cannot expel the abnormally thick mucous and infections make the worsen the situation. Opportunistic B. cepacia infection in patients with cystic fibrosis is life threatening and requires extensive chemotherapy as the organism is resistant to many antibiotics.
Most Commonly Found Signs And Symptoms In Cystic Fibrosis:
The signs and symptoms, age of onset and organs systems involved vary from person to person.
Diagnosis of the condition is bases on the following typical and atypical signs and symptoms and clinical
features:
1. Salty sweat (due to increased intracellular sodium)
2. Chronic cough with viscous sputum and hemoptysis (coughing up blood)
3. Nasal congestion
4. Recurrent respiratory infections
5. Chronic and persistent inflammation that causes bronchiectasis (permanent dilation and loss of contractility of the respiratory passage)
6. Obstruction of the bronchioles that leads to chronic obstructive pulmonary disease (COPD) like bronchitis and emphysema
7. Abdominal pain
8. Constipation with rectal prolapse
9. Pancreatitis
10. Diabetes mellitus (pancreatic pathology leads to destruction of pancreatic beta cells)
11. Weight loss (as a result of maldigestion and therefore malabsorption caused by obstruction of pancreatic enzymes in the ductal system)
12. Obstruction of the pancreatic duct with autolysis (destruction of pancreatic tissue by it’s own enzymes)
13. High infertility rate in men
What Causes Cystic Fibrosis:
Cystic fibrosis is inherited as an autosomal recessive disorder, both the mother and father should have the chronic disease for the child to be born with it. The chances of being born with the condition are 25% if both parents are “carriers” of the mutation and 75% if one parent has the active form of the disease and the second parent is only a carrier.

1. There are over 2000 mutations responsible for cystic fibrosis.
2. These mutations occur on the long arm of chromosome 7 and result in the formation of abnormal CFTR protein or the synthesis does not occur at all.
3. ΔF508 is the most common mutation.
The mutations are grouped into five classes:
1. Class 1: Mutations of protein synthesis
2. Class 2: Mutations of protein processing
3. Class 3: Mutations of gating
4. Class 4: Mutations of conduction
5. Class 5: Mutations with insufficient protein
Management And Treatment Of Cystic Fibrosis:
Genetic testing is performed for people who have the disease or are carriers to determine the chances of their offspring inheriting it. In case of lung disease, oral or systemic antibiotics are administered to combat infections and chest physiotherapy helps clear out mucous. Asthma may be treated with β-agonists and steroids. Lung transplantation and mechanical ventilators can be used in advanced disease. Vitamin supplements and pancreatic enzymes are used to treat maldigestion and malabsorption. Exogenous insulin is administered for those who develop type 2 diabetes mellitus. The combination of CFTR modulators “ivacaftor” and “lumacaftor” is also helpful in some patients. Various medical and surgical treatment options are available for infertility and ovulation issues.




